This web page was produced as an assignment for Genetics 564, an undergraduate capstone course at UW-Madison.
What is phylogenetics and what is a phylogenetic tree?
Phylogenetics is a branch of genetics which studies the evolutionary relationships between groups of organisms or taxa. A phylogenetic tree is a visual representation of these evolutionary relationships [1]. This tree can be constructed using DNA sequences, amino acid sequences of proteins, or structural/functional features. This allows researchers to examine the evolutionary history of specific features between organisms.
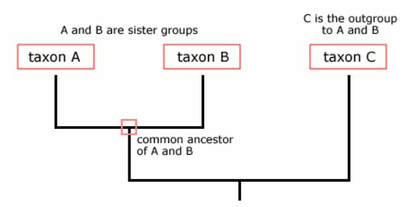
Figure 1. This figure shows the basic structure of a phylogenetic tree and its components. Each taxa is an individual organism, and nodes represent divergence from a common ancestor.
How are phylogenetic trees constructed?
Step 1: Sequence Alignment
First, the DNA, RNA, or protein sequence must be aligned. This step calculates differences between sequences and arranges them so conserved sections are together.
First, the DNA, RNA, or protein sequence must be aligned. This step calculates differences between sequences and arranges them so conserved sections are together.
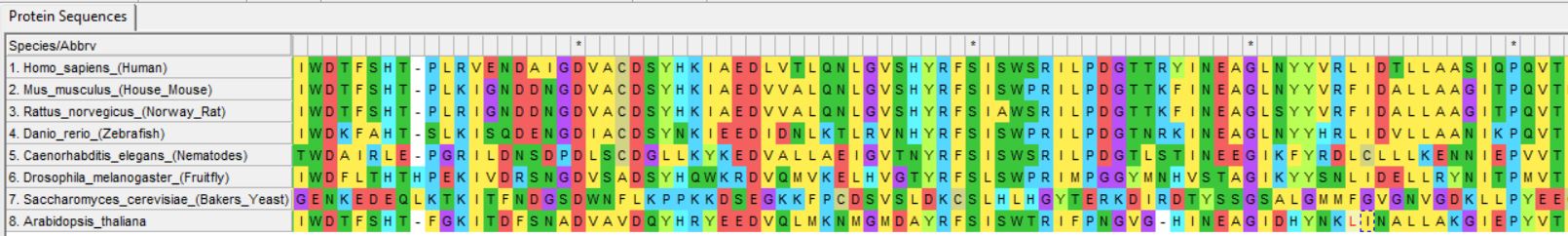
Figure 2. This figure shows a portion of the protein sequence (amino acids) alignment for the LCT gene homologs generated in MEGA.
Step 2: Construct Tree
There are many methods used for constructing phylogenetic trees but three commonly used methods are: neighbor joining, maximum likelihood, and maximum parsimony.
Neighbor Joining
Neighbor joining calculates pairwise differences between sequences. It then constructs a tree by calculating branch lengths based on the number of differences between two given sequences. It joins the two sequences that are most similar into one node, and repeats the process until all of the sequences have been added to the tree. This method uses physical distance is sequences as a representation for evolutionary distance.
Maximum Likelihood
Maximum likelihood is a statistic method to estimate parameters. In constructing trees, this method assigns probabilities to for groups of phylogenetic trees. It then determines a final tree by choosing the combination of most likely sub-trees. This method is useful for molecular genetics but can be computationally expensive.
Maximum Parsimony
This method constructs a tree by minimizing the total tree length. In this method the tree is constructed in a way that minimizes the number of changes or mutations required to get to the present day sequences, by examining all possible tree structures. This can be computationally expensive, because the number of possible trees grows quickly with the number of taxa included.
There are many methods used for constructing phylogenetic trees but three commonly used methods are: neighbor joining, maximum likelihood, and maximum parsimony.
Neighbor Joining
Neighbor joining calculates pairwise differences between sequences. It then constructs a tree by calculating branch lengths based on the number of differences between two given sequences. It joins the two sequences that are most similar into one node, and repeats the process until all of the sequences have been added to the tree. This method uses physical distance is sequences as a representation for evolutionary distance.
Maximum Likelihood
Maximum likelihood is a statistic method to estimate parameters. In constructing trees, this method assigns probabilities to for groups of phylogenetic trees. It then determines a final tree by choosing the combination of most likely sub-trees. This method is useful for molecular genetics but can be computationally expensive.
Maximum Parsimony
This method constructs a tree by minimizing the total tree length. In this method the tree is constructed in a way that minimizes the number of changes or mutations required to get to the present day sequences, by examining all possible tree structures. This can be computationally expensive, because the number of possible trees grows quickly with the number of taxa included.
Phylogenetic tree of Lactase
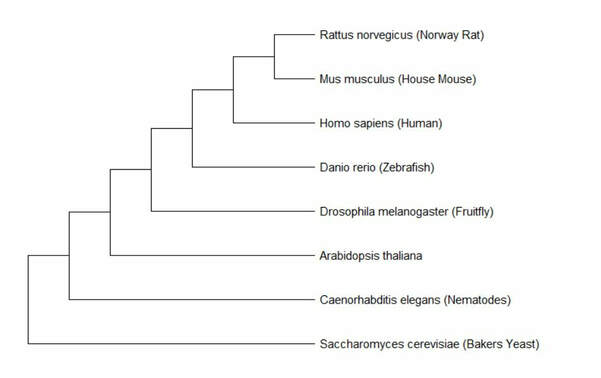
Figure 3. This is the tree constructed for the LCT protein homolog sequences using the neighbor joining method in MEGA.
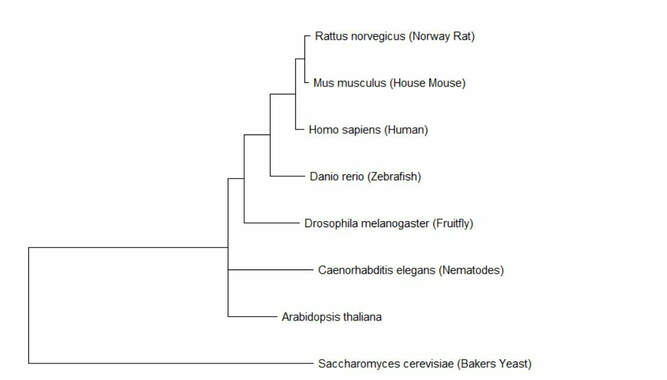
Figure 4. This is the phylogenetic tree constructed using the maximum likelihood method in MEGA.
Conclusions
Both tree making methods resulted in very similar phylogenetic trees. The only difference was that maximum likelihood had C. elegans and A. thaliana on one node together, while neighbor joining had each organism on its own node. These trees demonstrate that the rat and mouse homologs for lactase are most similar to humans, with the zebrafish being the next most similar. C. elegans, Arabidbopsis, and yeast were all much more distantly related. This conclusion follows the expected results from looking at the evolution of breastfeeding and the protein homologs.
References
[1] Understanding Evolution. Reading trees: A quick review. Berkley. Retrieved from evolution.berkeley.edu/evolibrary/article/phylogenetics_02
[2] Biro, R. (2015). Constructing Phylogenetic Trees. Retrieved from berkri.web.elte.hu/Theses/Biro.pdf
MEGA: Kumar, S. et al. (2018) Mega X: Molecular Evolutionary Genetics Analysis across computing platforms. Molecular Biology and Evolution. Retrieved from https://www.megasoftware.net/
[1] Understanding Evolution. Reading trees: A quick review. Berkley. Retrieved from evolution.berkeley.edu/evolibrary/article/phylogenetics_02
[2] Biro, R. (2015). Constructing Phylogenetic Trees. Retrieved from berkri.web.elte.hu/Theses/Biro.pdf
MEGA: Kumar, S. et al. (2018) Mega X: Molecular Evolutionary Genetics Analysis across computing platforms. Molecular Biology and Evolution. Retrieved from https://www.megasoftware.net/
